ZnGeP
2crystals are the frequency conversion materials with the excellent comprehensive performances in a range of 3–5 μm. However, the overlap of the absorption band and the pump wavelength range of optical parametric oscillator at 8–12 μm limits the application performance of the optical parametric oscillator and makes it impossible to achieve a far-infrared laser output. In this work, the formation energy and migration mechanism of six kinds of defects of ZnGeP
2crystal are discussed by density functional theory. The results show that two defective structures of
$\rm{V_P}$

and
$\rm{V_{Ge}}$

are difficult to form, while four defective structures of
$\rm V_{\rm Zn}^ -$

,
$\rm{Z{n_{Ge}}}$

,
$ {\rm Ge}_{\rm Zn}^ + $

and
$\rm{ G{e_{\rm Zn}} + {V_{\rm Zn}}}$

are easy to create. When the number of Ge atoms are slightly more than that of Zn atoms in ZnGeP
2crystals, the vacancy defects
$\rm V_{\rm Zn}^ -$

form more easily than antistructure defects
$ {\rm Ge}_{\rm Zn}^ + $
at 10 K, 500 K and 600 K, but the antistructure defects
$ {\rm Ge}_{\rm Zn}^ + $
are easier to form than the vacancy defects
$ {\text{V}}_{\text{Zn}}^{-} $

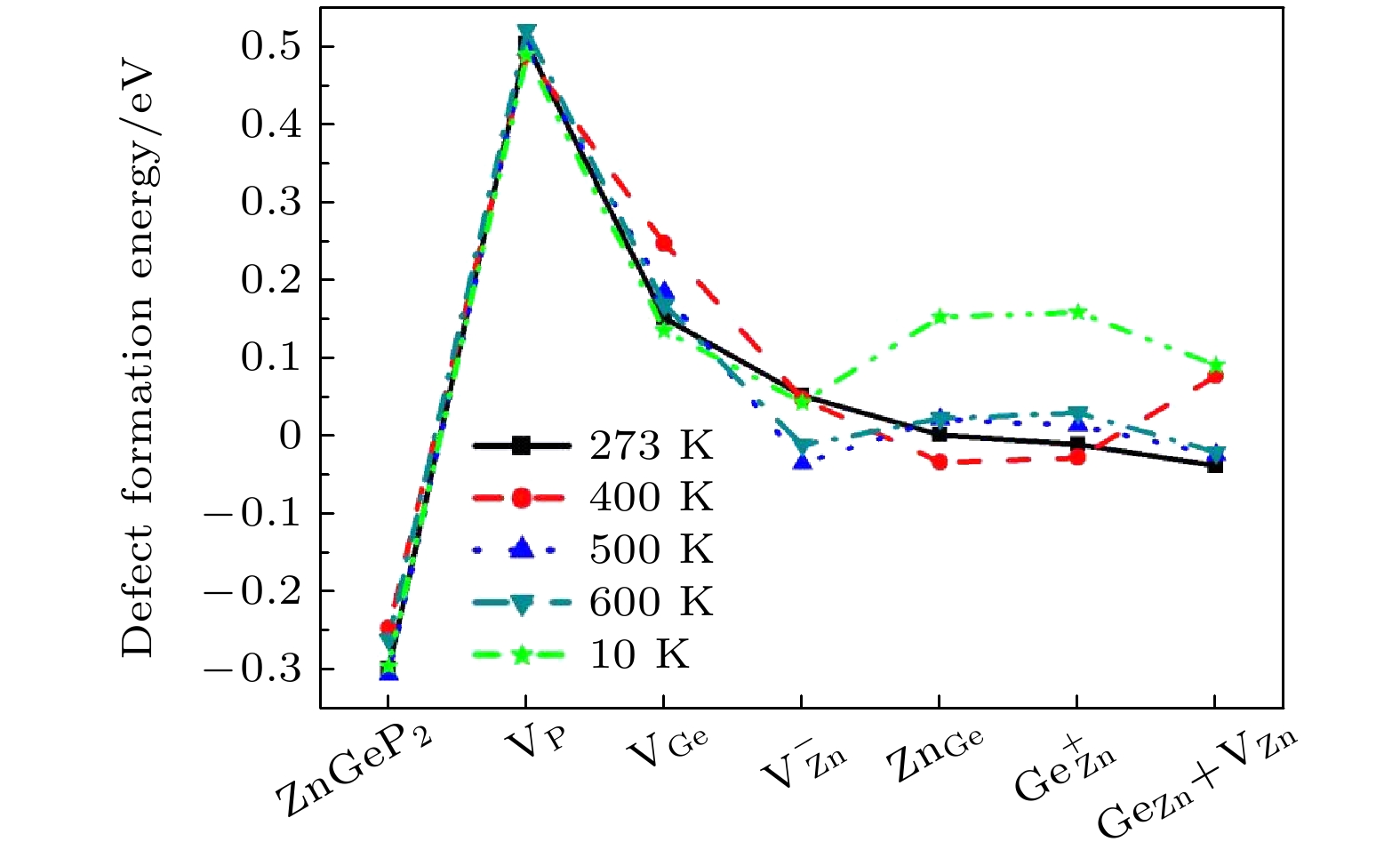
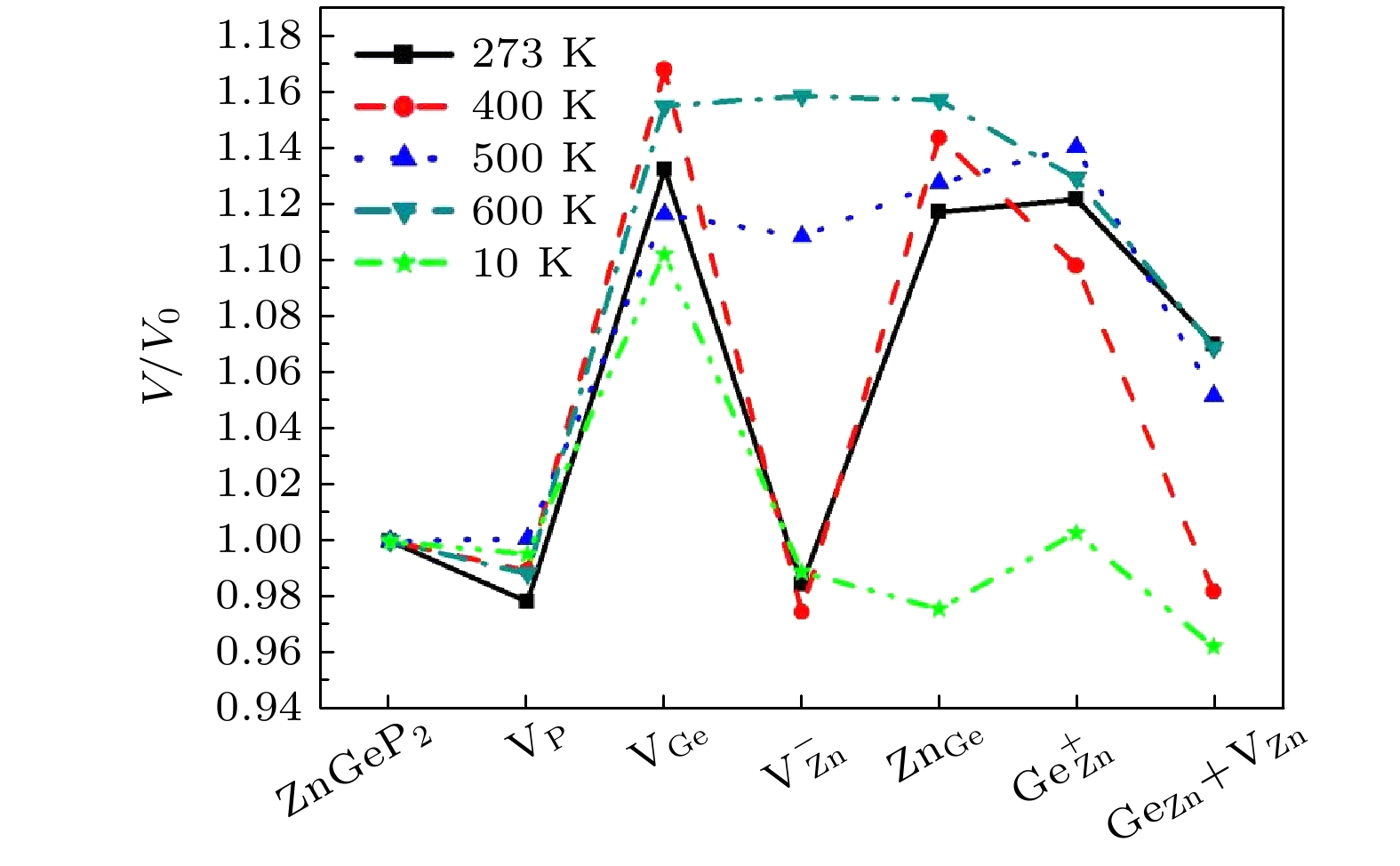
at 273 K and 400 K. There is a negative correlation between the volume expansion rate and the defect formation energy of ZnGeP
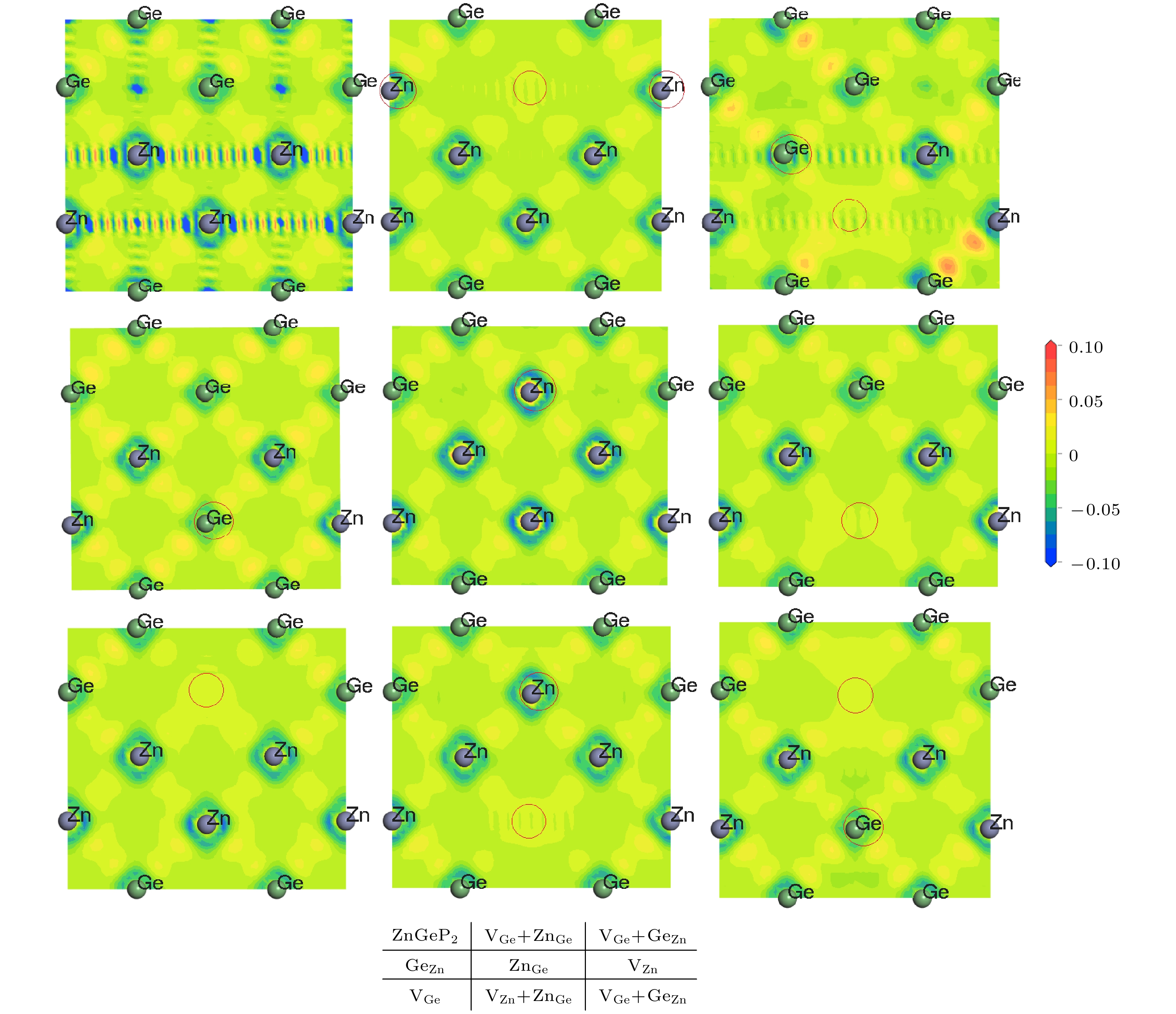
2crystal. The larger the volume expansion rate, the lower the defect formation energy is. The differential charge density shows that the electron cloud density among the atoms is enhanced in the defective structures of Ge
Znand V
Zn+Ge
Zn. The electron cloud density at the lattices of vacancy defects is enhanced when the vacancy defects (V
Znand V
Ge) and antistructure defects (Ge
Znand Zn
Ge) form the joint defects. Comparing with the defect-free cells, the charge of Zn atoms increases significantly, that of Ge is significantly reduced, and that of P does not change in the antistructure defect Zn
Geor Ge
Zn. The absorption spectra of ZnGeP
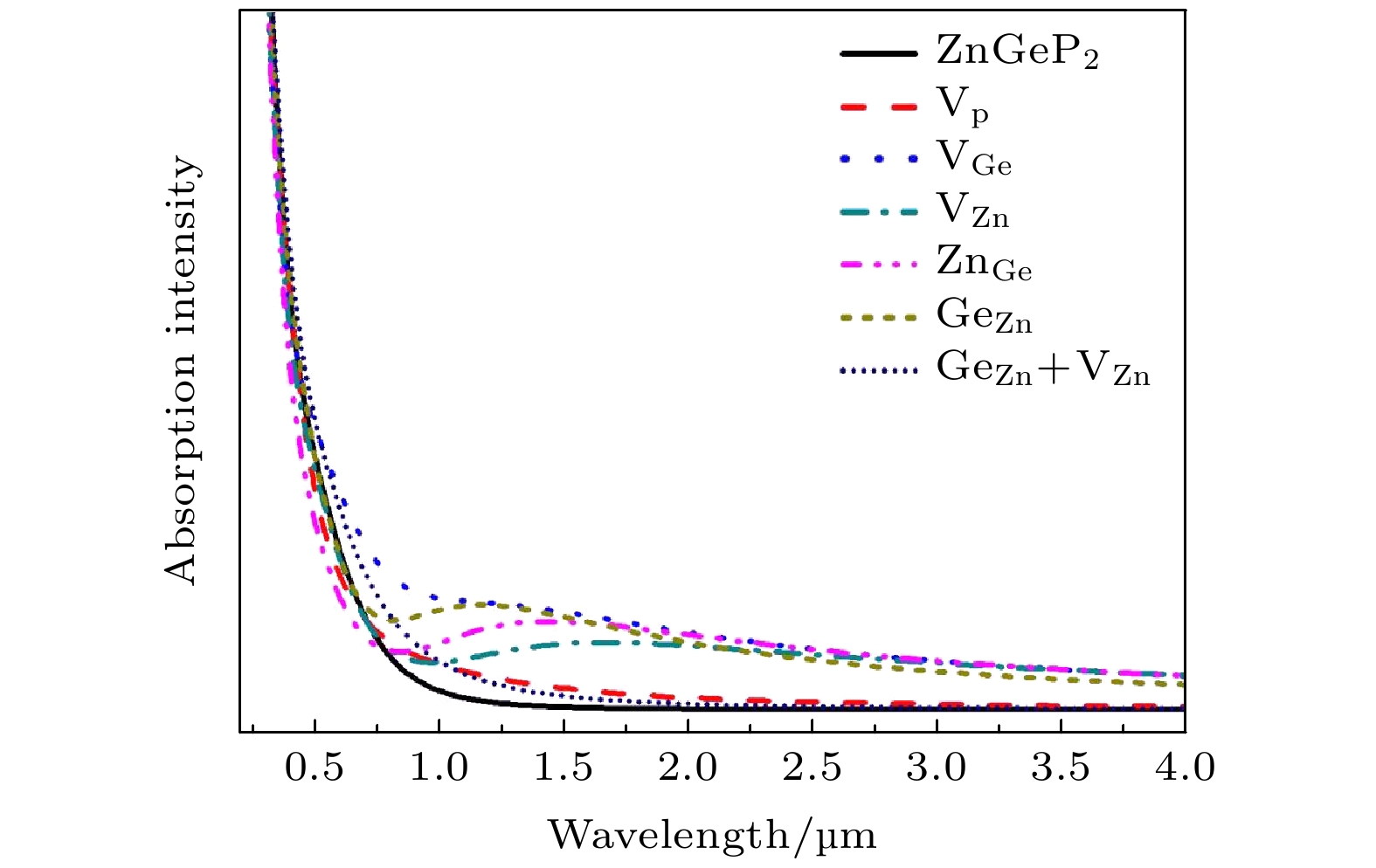
2crystal at 10K show that there is the significant absorption in a wavelength range from 0.6 μm to 2.5 μm for the four defective structures: V
Ge, V
Zn, Zn
Geand Ge
Zn, while the absorption in this range is small for the defective structures V
Pand Ge
Zn+V
Zn. The V
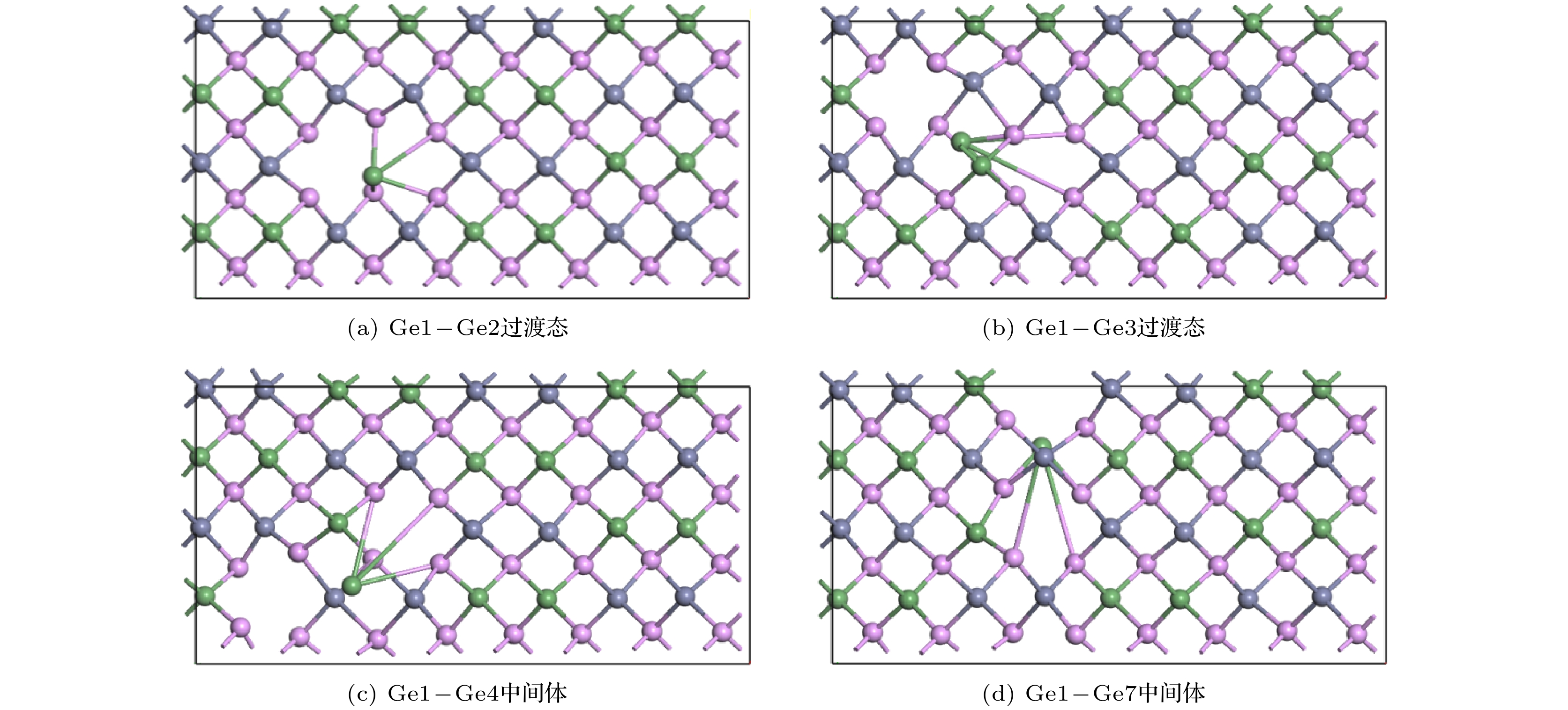
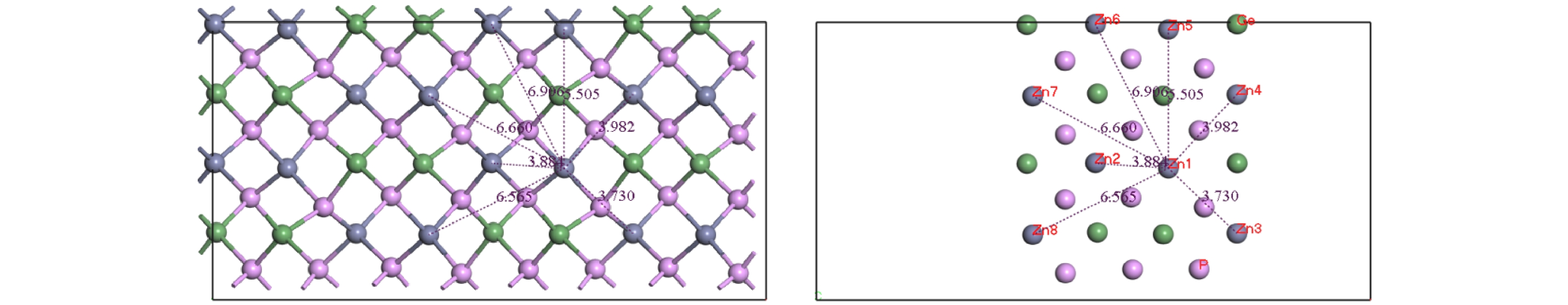
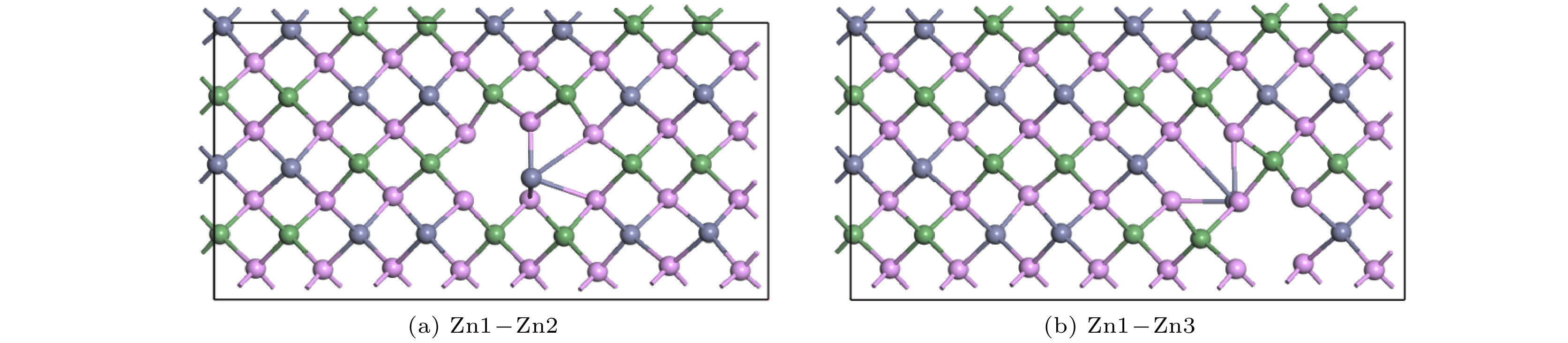
Znhas the lowest migration energy, while V
Gehas the highest. The difficulty for V
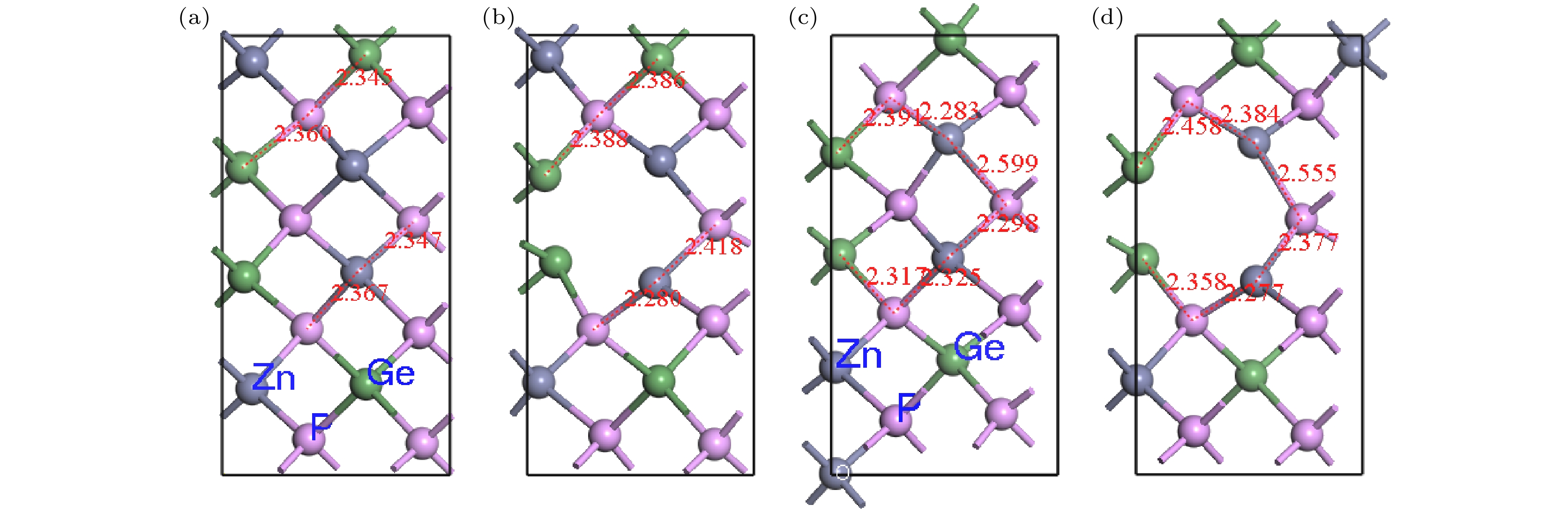
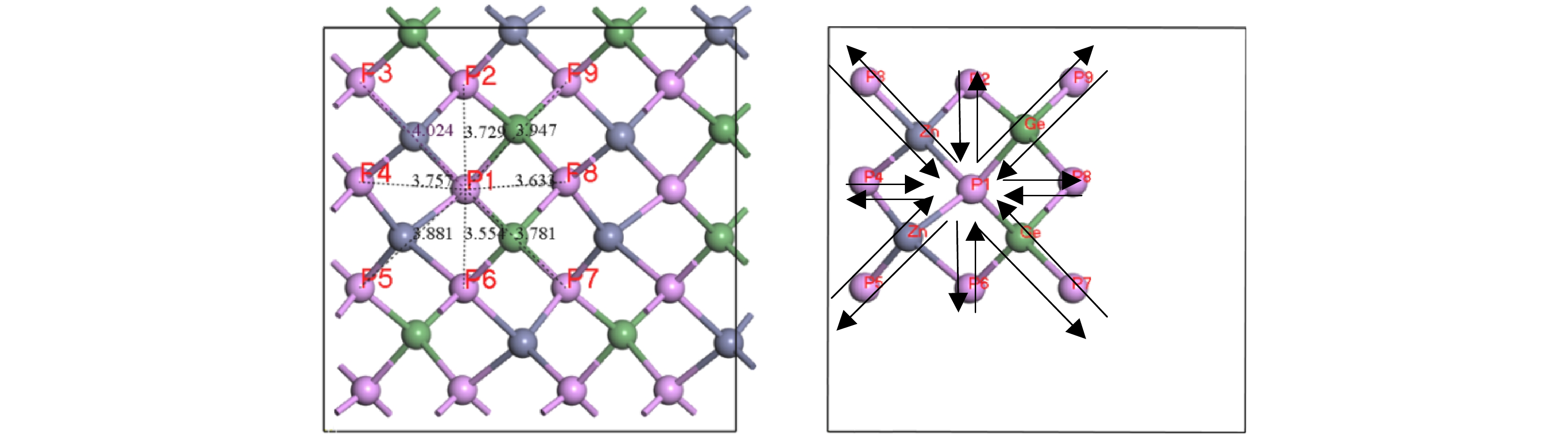
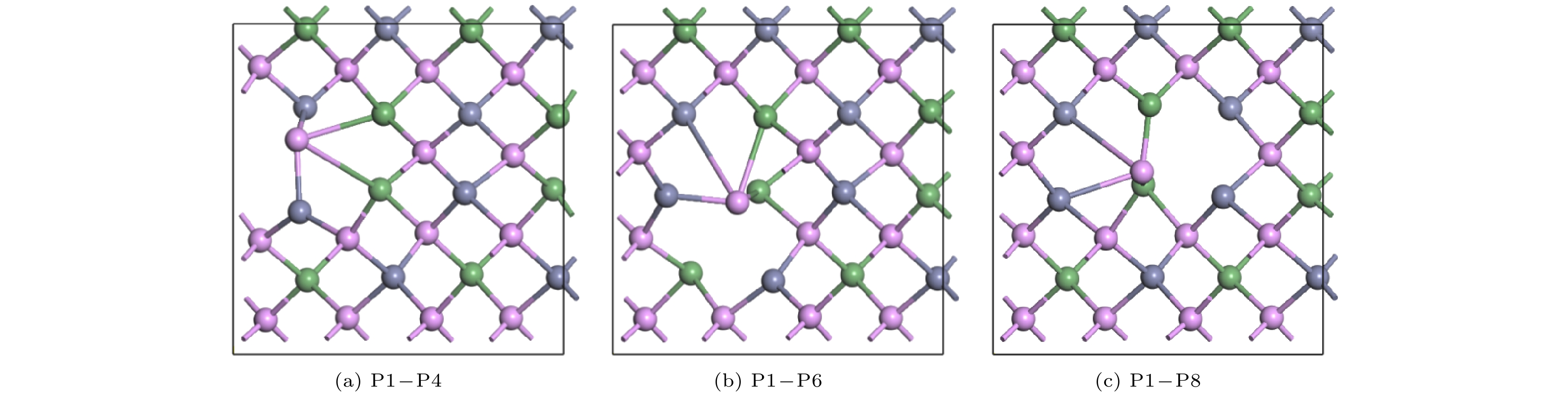
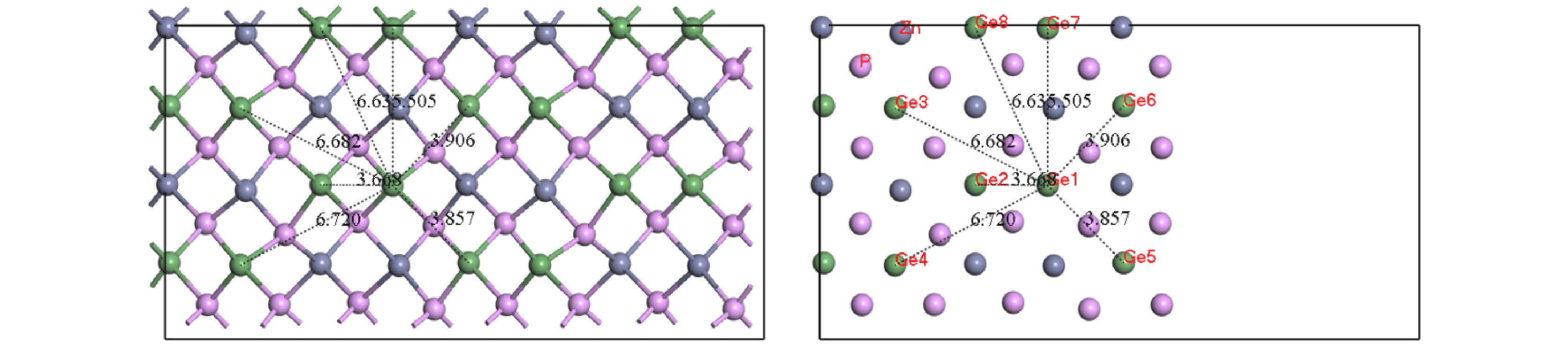
Pto migrate depends on the space resistance, while the difficulty for V
Geand V
Znto migrate depend on the inter-atomic distance. This may be related to the small radius and high proportion of P atoms and the large radius and low proportion of Ge and Zn atom in ZnGeP
2crystal.












 DownLoad:
CSV
DownLoad:
CSV










 DownLoad:
DownLoad:









