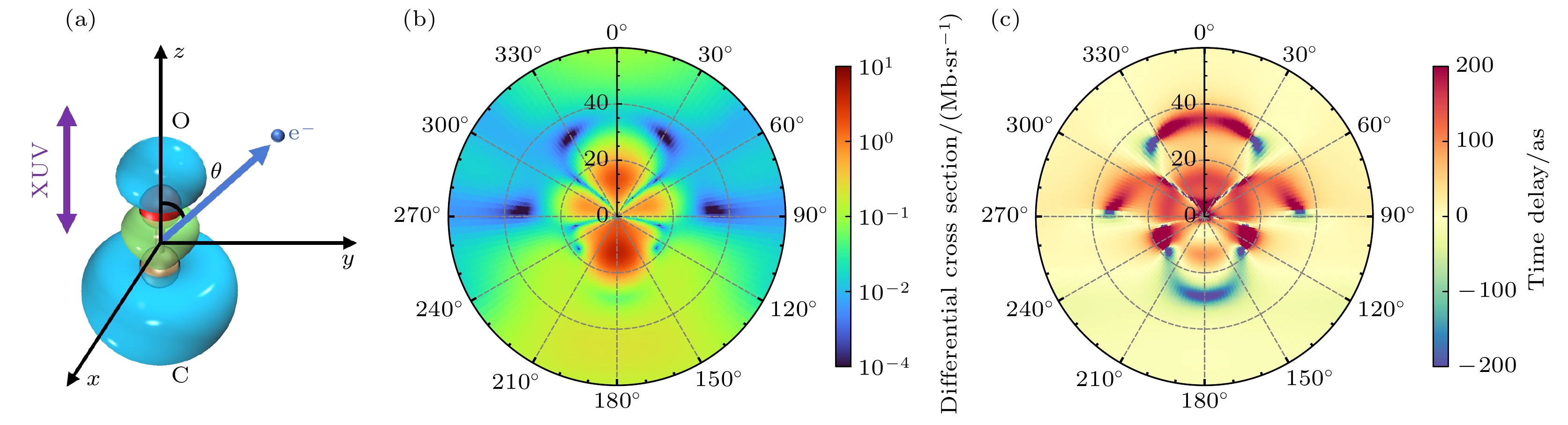
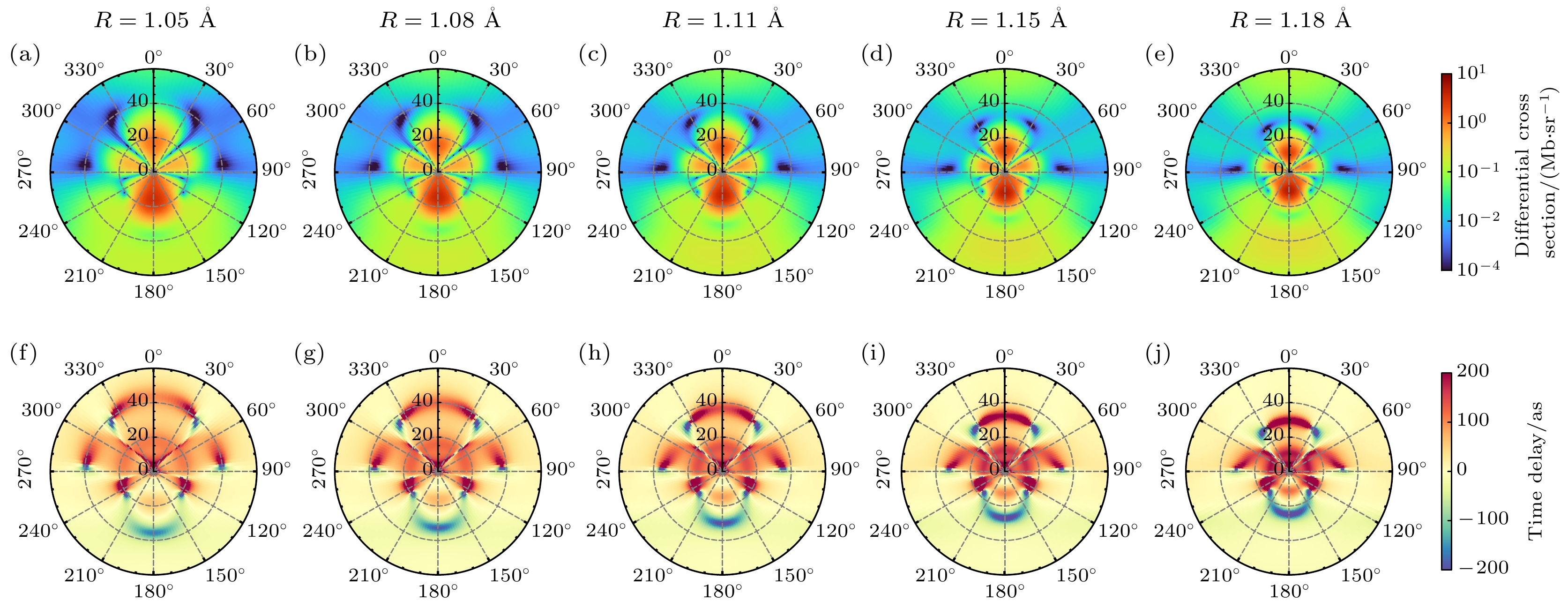
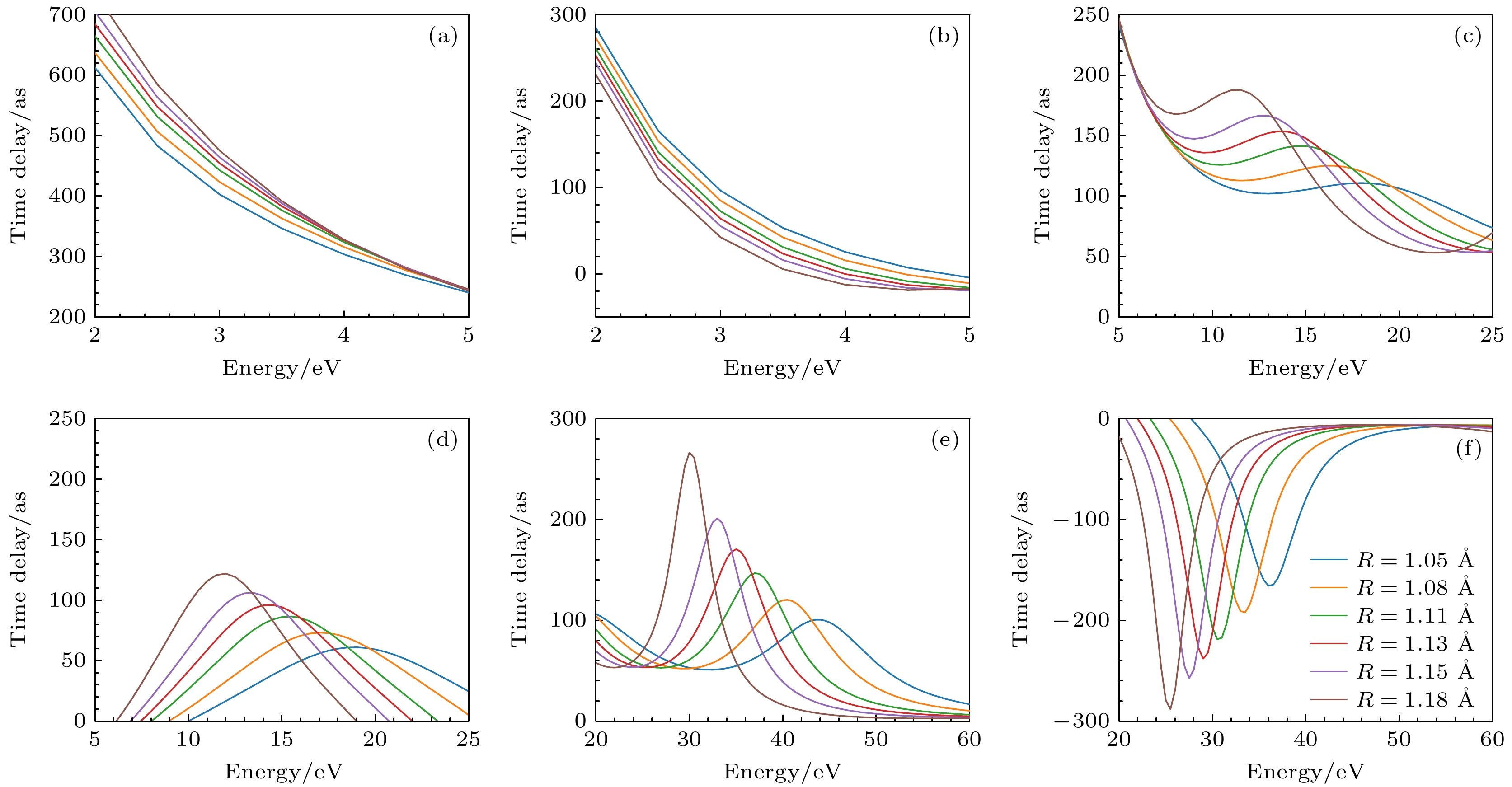
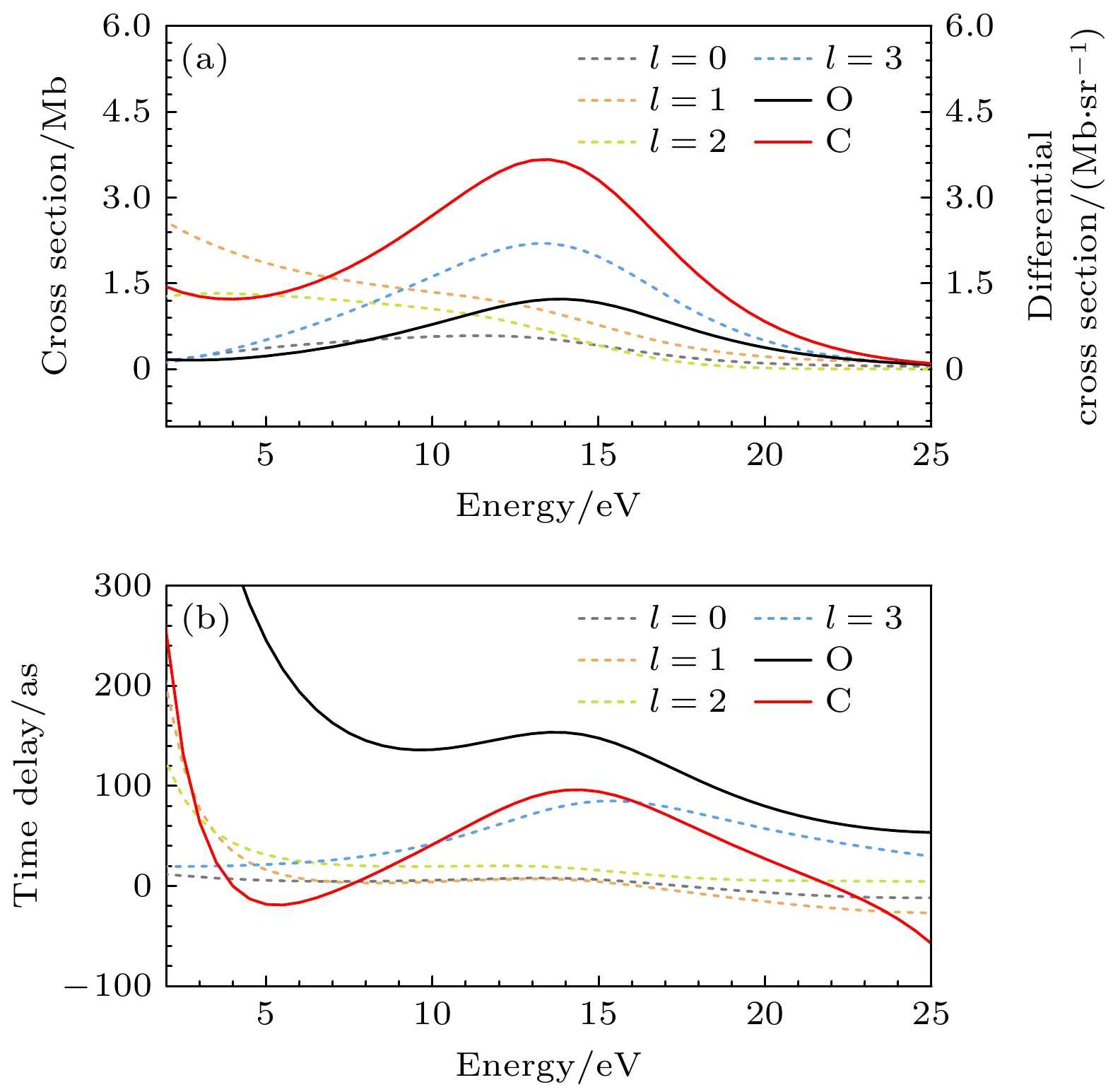
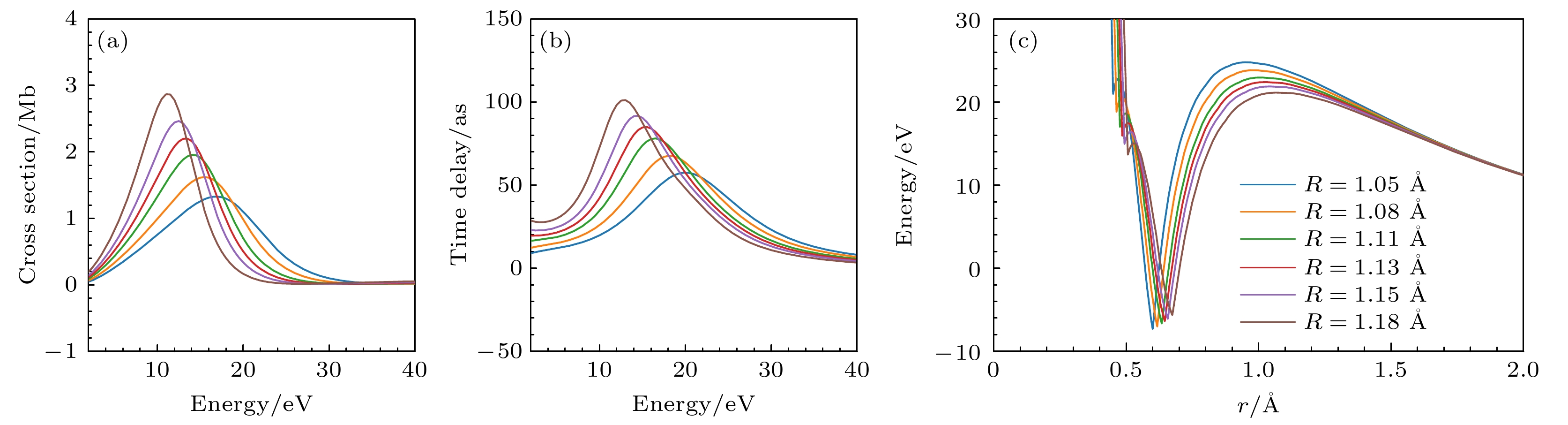
Photoionization time delay in atoms and molecules is a fundamental phenomenon in attosecond physics, encoding essential information about electronic structure and dynamics. Compared with atoms, molecules exhibit anisotropic potentials and additional nuclear degrees of freedom, which make the interpretation of molecular photoionization time delays more intricate but also more informative. In this work, we investigate the dependence of the photoionization time delay on the internuclear distance in the $ 5\sigma \to k\sigma$ ionization channel of carbon monoxide (CO) molecules. The molecular ground state is obtained using the Hartree-Fock method, and the photoionization process is treated within quantum scattering theory based on the iterative Schwinger variational principle of the Lippmann–Schwinger equation. Numerical calculations are performed with the ePolyScat program to obtain molecular-frame differential photoionization cross sections and time delays at various internuclear distances. Our results show that the extrema of the photoionization time delay occur near the peaks and dips of the differential cross section and shift toward lower energies as the internuclear distance R increases. At low energies, the time delay along the oxygen end increases with R, while that along the carbon end decreases, which is attributed to the asymmetric charge distribution and the resulting short-range potential difference between the two atomic sites. Around the shape-resonance energy region, both cross section and time delay display pronounced peaks associated with an $ l=3$ quasi-bound state. As R increases, the effective potential barrier broadens, the quasi-bound state energy moves to lower values, and its lifetime becomes longer, leading to enhanced resonance amplitude and increased time delay. In the high-energy region, opposite-sign peaks of time delay are found along the O and C directions, corresponding to minima in the cross section. These features are well explained by a two-center interference model, where increasing R shifts the interference minima and the associated time-delay peaks toward lower energies. This study provides deeper insights into the photoionization dynamics of CO molecules, accounting for the role of nuclear motion, and offers valuable references for studying the photoelectron dynamics of more complex molecular systems.














 DownLoad:
DownLoad:




